Packages for this tutorial
We will use the VariantAnnotation package from Bioconductor to read and filter VCFs. We will also use ape to make a phylogenetic tree. If you don’t have these packages already, install them like so:
install.packages("BiocManager", "ape")
BiocManager::install("VariantAnnotation")
Then load them.
library(VariantAnnotation)
library(ape)
Data for this tutorial
We will use the VCF that was generated during the GBS-SNP-CROP tutorial, which we named Msa_GSCdemo.vcf. It contains data for about 38K markers on eight tetraploid individuals of Miscanthus sacchariflorus.
It is generally easier to work with VCFs in VariantAnnotation if they have been zipped and indexed, so we’ll do that first.
mybg <- bgzip("Msa_GSCdemo.vcf")
indexTabix(mybg, format = "vcf")
We can take a look at some information from the file header.
hdr <- scanVcfHeader(mybg)
hdr
## class: VCFHeader
## samples(8): JM-2014-H-28 JM-2014-K-8 ... KMS257 RU2012-055
## meta(4): fileDate fileformat phasing source
## fixed(0):
## info(5): AC AF DP AV NS
## geno(2): GT AD
samples(hdr)
## [1] "JM-2014-H-28" "JM-2014-K-8" "JM-2014-M-5" "JM-2014-S-5"
## [5] "JY134" "JY185" "KMS257" "RU2012-055"
meta(hdr)$source
## DataFrame with 1 row and 1 column
## Value
## <character>
## source GBS-SNP-CROP
geno(hdr)
## DataFrame with 2 rows and 3 columns
## Number Type Description
## <character> <character> <character>
## GT 1 String Genotype
## AD R Integer Allele Depth
And confirm this matches the text in the file.
cat(readLines(mybg, 20), sep = "\n")
## ##fileformat=VCFv4.2
## ##fileDate=Thu Apr 25 10:55:57 2019
## ##source=GBS-SNP-CROP
## ##phasing=partial
## ##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele Count">
## ##INFO=<ID=AF,Number=A,Type=Integer,Description="Allele Frequency">
## ##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
## ##INFO=<ID=AV,Number=1,Type=Integer,Description="Average Depth">
## ##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data">
## ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
## ##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allele Depth">
## #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT JM-2014-H-28 JM-2014-K-8 JM-2014-M-5 JM-2014-S-5 JY134 JY185 KMS257 RU2012-055
## MockRefGenome 1731 . A G . PASS AC=8;AF=0.500;DP=7403;AV=925.38;NS=8 GT:AD 0/1:189,76 0/1:511,419 0/1:238,191 0/1:845,644 0/1:607,600 0/1:233,204 0/1:726,480 0/1:779,661
## MockRefGenome 1851 . T A . PASS AC=7;AF=0.438;DP=6180;AV=871.14;NS=7 GT:AD 0/1:646,167 0/1:720,102 0/1:240,48 0/1:520,103 0/1:898,188 ./.:.,. 0/1:679,170 0/1:1342,275
## MockRefGenome 3311 . A G . PASS AC=3;AF=0.188;DP=4447;AV=555.88;NS=8 GT:AD 0/0:23,0 0/0:349,0 0/0:464,0 0/1:521,165 0/1:620,121 0/1:311,104 0/0:898,0 0/0:871,0
## MockRefGenome 3833 . G T . PASS AC=8;AF=0.500;DP=4091;AV=511.38;NS=8 GT:AD 0/1:460,130 0/1:359,230 0/1:283,104 0/1:335,174 0/1:607,178 0/1:160,61 0/1:329,86 0/1:496,99
## MockRefGenome 5102 . T C . PASS AC=8;AF=0.500;DP=4108;AV=513.50;NS=8 GT:AD 0/1:203,154 0/1:299,118 0/1:181,52 0/1:616,209 0/1:524,191 0/1:253,58 0/1:445,210 0/1:440,155
## MockRefGenome 5104 . C G . PASS AC=8;AF=0.500;DP=4107;AV=513.38;NS=8 GT:AD 0/1:203,155 0/1:299,118 0/1:181,50 0/1:596,230 0/1:514,200 0/1:248,63 0/1:430,226 0/1:439,155
## MockRefGenome 5127 . G A . PASS AC=8;AF=0.500;DP=4107;AV=513.38;NS=8 GT:AD 0/1:203,155 0/1:298,118 0/1:181,52 0/1:595,230 0/1:514,200 0/1:245,63 0/1:431,227 0/1:440,155
## MockRefGenome 5327 . A G . PASS AC=8;AF=0.500;DP=3290;AV=411.25;NS=8 GT:AD 0/1:155,104 0/1:384,91 0/1:125,68 0/1:251,41 0/1:291,89 0/1:161,35 0/1:478,98 0/1:734,185
Making a prefilter
I know that Miscanthus has a whole genome duplication (even before the autotetraploidy in these M. sacchariflorus accessions) and so even with the paralog filtering that was performed by GBS-SNP-CROP, I’m suspicious that some paralogs made it through. Of those first few markers that I printed out, I can see that most of them are heterozygous for all individuals. Those markers are probably merged paralogs, and definitely not informative, so I would like to get rid of them.
One option from VariantAnnotation is to make a prefilter. This is a function that treats each line of the VCF genotype table as a text string, and returns TRUE or FALSE incidating whether or not that line should pass the filter. One very handy function for building prefilter functions is grepl, since it searches for a particular string or pattern, and returns TRUE if it finds it and FALSE otherwise. Another handy function is gregexpr if you want to know how many times you find a particular pattern in each line.
If all eight samples are either heterozygous or missing, I want to discard the marker. In the sense of the file format, this means that the GT field for every sample is either 0/1 or ./.. Another way of looking at it is that there would be no homozygotes, i.e. no 0/0 and no 1/1. Here’s a function to perform that check.
NotAllHet <- function(lines){
found00 <- grepl("0/0:", lines)
found11 <- grepl("1/1:", lines)
return(found00 | found11)
}
We can test it with the first few lines of the file, ignoring the header lines.
mylines <- readLines(mybg, 30)
# skip header lines starting with "#"
mylines <- mylines[!grepl("^#", mylines)]
keepem <- NotAllHet(mylines)
cat(mylines[keepem], sep = "\n")
## MockRefGenome 3311 . A G . PASS AC=3;AF=0.188;DP=4447;AV=555.88;NS=8 GT:AD 0/0:23,0 0/0:349,0 0/0:464,0 0/1:521,165 0/1:620,121 0/1:311,104 0/0:898,0 0/0:871,0
## MockRefGenome 6284 . G A . PASS AC=5;AF=0.312;DP=3185;AV=398.00;NS=8 GT:AD 0/0:292,1 0/0:713,0 0/1:235,60 0/1:474,172 0/1:234,219 0/1:124,128 0/1:153,44 0/0:336,0
## MockRefGenome 6333 . A T . PASS AC=5;AF=0.312;DP=3169;AV=396.12;NS=8 GT:AD 0/0:291,0 0/0:711,0 0/1:223,69 0/1:437,203 0/1:234,219 0/1:125,126 0/1:118,78 0/0:335,0
## MockRefGenome 7285 . C T . PASS AC=3;AF=0.188;DP=3123;AV=423.43;NS=7 GT:AD 0/0:388,0 0/0:552,0 ./.:.,. 0/1:372,98 0/1:489,334 0/1:138,123 0/0:235,1 0/0:235,0
cat(mylines[!keepem], sep = "\n")
## MockRefGenome 1731 . A G . PASS AC=8;AF=0.500;DP=7403;AV=925.38;NS=8 GT:AD 0/1:189,76 0/1:511,419 0/1:238,191 0/1:845,644 0/1:607,600 0/1:233,204 0/1:726,480 0/1:779,661
## MockRefGenome 1851 . T A . PASS AC=7;AF=0.438;DP=6180;AV=871.14;NS=7 GT:AD 0/1:646,167 0/1:720,102 0/1:240,48 0/1:520,103 0/1:898,188 ./.:.,. 0/1:679,170 0/1:1342,275
## MockRefGenome 3833 . G T . PASS AC=8;AF=0.500;DP=4091;AV=511.38;NS=8 GT:AD 0/1:460,130 0/1:359,230 0/1:283,104 0/1:335,174 0/1:607,178 0/1:160,61 0/1:329,86 0/1:496,99
## MockRefGenome 5102 . T C . PASS AC=8;AF=0.500;DP=4108;AV=513.50;NS=8 GT:AD 0/1:203,154 0/1:299,118 0/1:181,52 0/1:616,209 0/1:524,191 0/1:253,58 0/1:445,210 0/1:440,155
## MockRefGenome 5104 . C G . PASS AC=8;AF=0.500;DP=4107;AV=513.38;NS=8 GT:AD 0/1:203,155 0/1:299,118 0/1:181,50 0/1:596,230 0/1:514,200 0/1:248,63 0/1:430,226 0/1:439,155
## MockRefGenome 5127 . G A . PASS AC=8;AF=0.500;DP=4107;AV=513.38;NS=8 GT:AD 0/1:203,155 0/1:298,118 0/1:181,52 0/1:595,230 0/1:514,200 0/1:245,63 0/1:431,227 0/1:440,155
## MockRefGenome 5327 . A G . PASS AC=8;AF=0.500;DP=3290;AV=411.25;NS=8 GT:AD 0/1:155,104 0/1:384,91 0/1:125,68 0/1:251,41 0/1:291,89 0/1:161,35 0/1:478,98 0/1:734,185
## MockRefGenome 5498 . A C . PASS AC=7;AF=0.438;DP=3112;AV=400.14;NS=7 GT:AD ./.:.,. 0/1:344,71 0/1:96,53 0/1:338,95 0/1:418,91 0/1:247,81 0/1:279,80 0/1:481,127
## MockRefGenome 6514 . G C . PASS AC=7;AF=0.438;DP=3970;AV=566.86;NS=7 GT:AD 0/1:360,189 0/1:323,121 0/1:122,111 0/1:227,89 0/1:478,177 ./.:.,. 0/1:337,246 0/1:738,450
## MockRefGenome 6832 . A T . PASS AC=7;AF=0.438;DP=10029;AV=1204.14;NS=7 GT:AD 0/1:1479,226 0/1:1216,210 0/1:641,105 ./.:.,. 0/1:651,87 0/1:325,55 0/1:1333,238 0/1:1580,283
## MockRefGenome 6835 . G T . PASS AC=7;AF=0.438;DP=9750;AV=1339.14;NS=7 GT:AD 0/1:1447,240 0/1:1127,269 0/1:602,112 0/1:1290,274 0/1:531,154 ./.:.,. 0/1:1289,222 0/1:1546,271
## MockRefGenome 7510 . C A . PASS AC=8;AF=0.500;DP=2853;AV=356.62;NS=8 GT:AD 0/1:203,67 0/1:262,77 0/1:131,48 0/1:222,102 0/1:515,111 0/1:225,57 0/1:407,71 0/1:253,102
## MockRefGenome 8478 . C G . PASS AC=8;AF=0.500;DP=4276;AV=534.50;NS=8 GT:AD 0/1:363,115 0/1:493,131 0/1:196,84 0/1:317,104 0/1:562,122 0/1:253,44 0/1:439,128 0/1:756,169
## MockRefGenome 8490 . G C . PASS AC=8;AF=0.500;DP=4272;AV=534.00;NS=8 GT:AD 0/1:251,227 0/1:368,254 0/1:154,126 0/1:256,165 0/1:398,286 0/1:206,91 0/1:346,220 0/1:614,310
Here is how I would apply that filter to my VCF file, exporting a filtered VCF.
filter1_file <- "Msa_GSCdemo_filter1.vcf"
filterVcf(mybg, genome = "MsaMockReference",
destination = filter1_file,
prefilters = FilterRules(list(NotAllHet)))
And I can confirm that it looks filtered.
cat(readLines(filter1_file, 20), sep = "\n")
## ##fileformat=VCFv4.2
## ##fileDate=Thu Apr 25 10:55:57 2019
## ##source=GBS-SNP-CROP
## ##phasing=partial
## ##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele Count">
## ##INFO=<ID=AF,Number=A,Type=Integer,Description="Allele Frequency">
## ##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
## ##INFO=<ID=AV,Number=1,Type=Integer,Description="Average Depth">
## ##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data">
## ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
## ##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allele Depth">
## #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT JM-2014-H-28 JM-2014-K-8 JM-2014-M-5 JM-2014-S-5 JY134 JY185 KMS257 RU2012-055
## MockRefGenome 3311 . A G . PASS AC=3;AF=0.188;DP=4447;AV=555.88;NS=8 GT:AD 0/0:23,0 0/0:349,0 0/0:464,0 0/1:521,165 0/1:620,121 0/1:311,104 0/0:898,0 0/0:871,0
## MockRefGenome 6284 . G A . PASS AC=5;AF=0.312;DP=3185;AV=398.00;NS=8 GT:AD 0/0:292,1 0/0:713,0 0/1:235,60 0/1:474,172 0/1:234,219 0/1:124,128 0/1:153,44 0/0:336,0
## MockRefGenome 6333 . A T . PASS AC=5;AF=0.312;DP=3169;AV=396.12;NS=8 GT:AD 0/0:291,0 0/0:711,0 0/1:223,69 0/1:437,203 0/1:234,219 0/1:125,126 0/1:118,78 0/0:335,0
## MockRefGenome 7285 . C T . PASS AC=3;AF=0.188;DP=3123;AV=423.43;NS=7 GT:AD 0/0:388,0 0/0:552,0 ./.:.,. 0/1:372,98 0/1:489,334 0/1:138,123 0/0:235,1 0/0:235,0
## MockRefGenome 8822 . T A . PASS AC=4;AF=0.250;DP=5573;AV=718.86;NS=7 GT:AD 0/0:264,0 ./.:.,. 0/1:300,63 0/1:670,87 0/0:963,0 0/1:199,29 0/0:872,0 0/1:1308,277
## MockRefGenome 11589 . C A . PASS AC=3;AF=0.188;DP=3951;AV=493.88;NS=8 GT:AD 0/1:281,103 0/1:429,334 0/1:202,109 0/0:541,0 0/0:583,0 0/0:225,0 0/0:576,0 0/0:568,0
## MockRefGenome 12355 . C T . PASS AC=2;AF=0.125;DP=1681;AV=222.00;NS=7 GT:AD 0/0:287,1 0/1:215,55 0/0:57,0 0/1:176,32 0/0:169,0 ./.:.,. 0/0:263,0 0/0:300,0
## MockRefGenome 12663 . T C . PASS AC=2;AF=0.125;DP=1807;AV=226.29;NS=7 GT:AD 0/0:118,0 ./.:.,. 0/0:76,0 0/1:327,192 0/0:270,0 0/1:50,9 0/0:223,0 0/0:319,0
Then zip and index it.
filter1_bg <- bgzip(filter1_file)
indexTabix(filter1_bg, format = "vcf")
Importing data from a VCF
This is a pretty small VCF, so let’s first explore what happens when we import the whole thing.
all_my_data <- readVcf(filter1_bg)
all_my_data
## class: CollapsedVCF
## dim: 29294 8
## rowRanges(vcf):
## GRanges with 5 metadata columns: paramRangeID, REF, ALT, QUAL, FILTER
## info(vcf):
## DataFrame with 5 columns: AC, AF, DP, AV, NS
## info(header(vcf)):
## Number Type Description
## AC A Integer Allele Count
## AF A Integer Allele Frequency
## DP 1 Integer Total Depth
## AV 1 Integer Average Depth
## NS 1 Integer Number of Samples With Data
## geno(vcf):
## SimpleList of length 2: GT, AD
## geno(header(vcf)):
## Number Type Description
## GT 1 String Genotype
## AD R Integer Allele Depth
We kept 29K markers (pretty good given how heavily filtered the first few lines were). We also see a bunch of slots in the VCF object that we can explore.
We can get to the header, with the same data we found using scanVcfHeader on the file.
header(all_my_data)
## class: VCFHeader
## samples(8): JM-2014-H-28 JM-2014-K-8 ... KMS257 RU2012-055
## meta(4): fileDate fileformat phasing source
## fixed(0):
## info(5): AC AF DP AV NS
## geno(2): GT AD
Here’s the data from the leftmost columns of the genotype table. This is a GRanges object, which is a special kind of table used in Bioconductor for representing genomic coordinates.
rowRanges(all_my_data)
## GRanges object with 29294 ranges and 5 metadata columns:
## seqnames ranges strand | paramRangeID
## <Rle> <IRanges> <Rle> | <factor>
## MockRefGenome:3311_A/G MockRefGenome 3311 * | <NA>
## MockRefGenome:6284_G/A MockRefGenome 6284 * | <NA>
## MockRefGenome:6333_A/T MockRefGenome 6333 * | <NA>
## MockRefGenome:7285_C/T MockRefGenome 7285 * | <NA>
## MockRefGenome:8822_T/A MockRefGenome 8822 * | <NA>
## ... ... ... ... . ...
## MockRefGenome:39977669_A/G MockRefGenome 39977669 * | <NA>
## MockRefGenome:40101335_G/T MockRefGenome 40101335 * | <NA>
## MockRefGenome:40259299_T/C MockRefGenome 40259299 * | <NA>
## MockRefGenome:40949663_A/T MockRefGenome 40949663 * | <NA>
## MockRefGenome:40949695_A/G MockRefGenome 40949695 * | <NA>
## REF ALT QUAL
## <DNAStringSet> <DNAStringSetList> <numeric>
## MockRefGenome:3311_A/G A G <NA>
## MockRefGenome:6284_G/A G A <NA>
## MockRefGenome:6333_A/T A T <NA>
## MockRefGenome:7285_C/T C T <NA>
## MockRefGenome:8822_T/A T A <NA>
## ... ... ... ...
## MockRefGenome:39977669_A/G A G <NA>
## MockRefGenome:40101335_G/T G T <NA>
## MockRefGenome:40259299_T/C T C <NA>
## MockRefGenome:40949663_A/T A T <NA>
## MockRefGenome:40949695_A/G A G <NA>
## FILTER
## <character>
## MockRefGenome:3311_A/G PASS
## MockRefGenome:6284_G/A PASS
## MockRefGenome:6333_A/T PASS
## MockRefGenome:7285_C/T PASS
## MockRefGenome:8822_T/A PASS
## ... ...
## MockRefGenome:39977669_A/G PASS
## MockRefGenome:40101335_G/T PASS
## MockRefGenome:40259299_T/C PASS
## MockRefGenome:40949663_A/T PASS
## MockRefGenome:40949695_A/G PASS
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
The INFO column from the VCF also got unpacked into its own table.
info(all_my_data)
## DataFrame with 29294 rows and 5 columns
## AC AF DP AV
## <IntegerList> <IntegerList> <integer> <integer>
## MockRefGenome:3311_A/G 3 0 4447 555
## MockRefGenome:6284_G/A 5 0 3185 398
## MockRefGenome:6333_A/T 5 0 3169 396
## MockRefGenome:7285_C/T 3 0 3123 423
## MockRefGenome:8822_T/A 4 0 5573 718
## ... ... ... ... ...
## MockRefGenome:39977669_A/G 2 0 235 29
## MockRefGenome:40101335_G/T 2 0 198 27
## MockRefGenome:40259299_T/C 6 0 303 41
## MockRefGenome:40949663_A/T 10 0 353 44
## MockRefGenome:40949695_A/G 8 0 251 31
## NS
## <integer>
## MockRefGenome:3311_A/G 8
## MockRefGenome:6284_G/A 8
## MockRefGenome:6333_A/T 8
## MockRefGenome:7285_C/T 7
## MockRefGenome:8822_T/A 7
## ... ...
## MockRefGenome:39977669_A/G 8
## MockRefGenome:40101335_G/T 7
## MockRefGenome:40259299_T/C 7
## MockRefGenome:40949663_A/T 8
## MockRefGenome:40949695_A/G 8
We can get the definitions of those columns from the header.
info(header(all_my_data))
## DataFrame with 5 rows and 3 columns
## Number Type Description
## <character> <character> <character>
## AC A Integer Allele Count
## AF A Integer Allele Frequency
## DP 1 Integer Total Depth
## AV 1 Integer Average Depth
## NS 1 Integer Number of Samples With Data
Last but certainly not least, there are the data from the genotypes table. This includes strings representing the genotypes (GT) and allelic read depths (AD).
geno(all_my_data)$GT[1:10,]
## JM-2014-H-28 JM-2014-K-8 JM-2014-M-5 JM-2014-S-5
## MockRefGenome:3311_A/G "0/0" "0/0" "0/0" "0/1"
## MockRefGenome:6284_G/A "0/0" "0/0" "0/1" "0/1"
## MockRefGenome:6333_A/T "0/0" "0/0" "0/1" "0/1"
## MockRefGenome:7285_C/T "0/0" "0/0" "./." "0/1"
## MockRefGenome:8822_T/A "0/0" "./." "0/1" "0/1"
## MockRefGenome:11589_C/A "0/1" "0/1" "0/1" "0/0"
## MockRefGenome:12355_C/T "0/0" "0/1" "0/0" "0/1"
## MockRefGenome:12663_T/C "0/0" "./." "0/0" "0/1"
## MockRefGenome:13346_C/A "0/0" "0/1" "0/0" "0/1"
## MockRefGenome:13563_C/T "0/0" "0/0" "0/0" "0/0"
## JY134 JY185 KMS257 RU2012-055
## MockRefGenome:3311_A/G "0/1" "0/1" "0/0" "0/0"
## MockRefGenome:6284_G/A "0/1" "0/1" "0/1" "0/0"
## MockRefGenome:6333_A/T "0/1" "0/1" "0/1" "0/0"
## MockRefGenome:7285_C/T "0/1" "0/1" "0/0" "0/0"
## MockRefGenome:8822_T/A "0/0" "0/1" "0/0" "0/1"
## MockRefGenome:11589_C/A "0/0" "0/0" "0/0" "0/0"
## MockRefGenome:12355_C/T "0/0" "./." "0/0" "0/0"
## MockRefGenome:12663_T/C "0/0" "0/1" "0/0" "0/0"
## MockRefGenome:13346_C/A "./." "0/0" "0/1" "0/0"
## MockRefGenome:13563_C/T "./." "0/0" "0/1" "0/1"
geno(all_my_data)$AD[1:10,]
## JM-2014-H-28 JM-2014-K-8 JM-2014-M-5 JM-2014-S-5
## MockRefGenome:3311_A/G Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:6284_G/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:6333_A/T Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:7285_C/T Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:8822_T/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:11589_C/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:12355_C/T Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:12663_T/C Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:13346_C/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:13563_C/T Integer,2 Integer,2 Integer,2 Integer,2
## JY134 JY185 KMS257 RU2012-055
## MockRefGenome:3311_A/G Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:6284_G/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:6333_A/T Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:7285_C/T Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:8822_T/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:11589_C/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:12355_C/T Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:12663_T/C Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:13346_C/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:13563_C/T Integer,2 Integer,2 Integer,2 Integer,2
geno(all_my_data)$AD[[1,1]]
## [1] 23 0
This is about the stage where I’d like to do a sanity check to confirm that my samples show the pattern of relatedness that I’m expecting. I will convert the diploidized genotypes to numbers and make a neighbor-joining tree.
numgen <- matrix(NA_integer_, nrow = dim(all_my_data)[1],
ncol = dim(all_my_data)[2],
dimnames = dimnames(geno(all_my_data)$GT))
numgen[geno(all_my_data)$GT == "0/0"] <- 0L
numgen[geno(all_my_data)$GT == "0/1"] <- 1L
numgen[geno(all_my_data)$GT == "1/1"] <- 2L
mydist <- dist(t(numgen))
mynj <- nj(mydist)
plot(mynj, type = "unrooted")
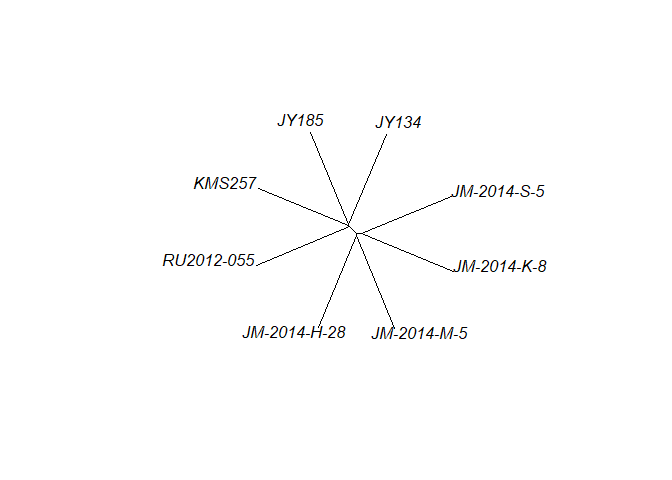
We can see a clustering of Japan vs. the mainland, and northern vs. southern Japan, as expected.
Making a filter
Before, we did prefiltering based on text processing alone. However, we can also make functions to filter the data after it has been imported to R.
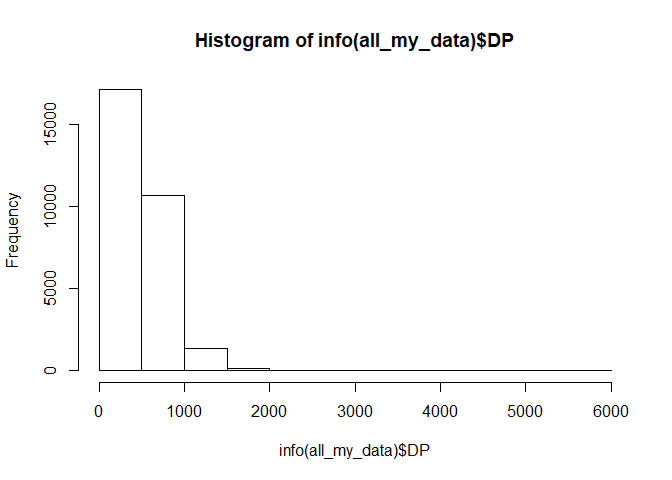
We had some rough cutoffs for depth using GBS-SNP-CROP, but we can narrow those further. What is the distribution of total depth among markers in the dataset?
hist(info(all_my_data)$DP)
hist(log(info(all_my_data)$DP))
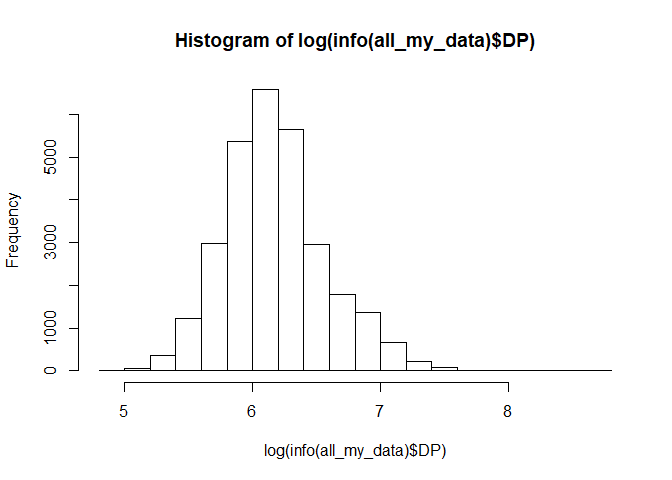
How about we say we want markers with a total depth below 1000. We can make a filter function for that.
DPbelow1000 <- function(vcf){
return(info(vcf)$DP < 1000)
}
mean(DPbelow1000(all_my_data))
## [1] 0.9479416
94.8% of markers would pass that threshold.
We also allowed 25% missing data when we ran GBS-SNP-CROP. We can see that when we look at NS, the number of samples with data.
table(info(all_my_data)$NS)
##
## 7 8
## 16473 12821
Or perhaps it was <25% missing data. If we wanted no missing data at all, we could make a filter function.
NoMissing <- function(vcf){
return(info(vcf)$NS == 8)
}
mean(NoMissing(all_my_data))
## [1] 0.4376664
Then we could use both of those functions as filters. We could even combine them with the prefilter we made before.
filter2_file <- "Msa_GSCdemo_filter2.vcf"
filterVcf(mybg, genome = "MsaMockReference",
destination = filter2_file,
prefilters = FilterRules(list(NotAllHet)),
filters = FilterRules(list(DPbelow1000, NoMissing)))
Working with big VCFs
That’s all well and good for this little 5 Mb VCF, where we could import the whole thing and explore it before deciding how we wanted to filter it. What about a 50 Gb VCF?
In VariantAnnotation, we can create a TabixFile object, which contains a pointer to the VCF file that we’re working with. When we do that, we can set yieldSize to indicate how many SNPs to import at once. This TabixFile object is a type of file connection.
mycon <- TabixFile(mybg, yieldSize = 1000)
We open the connection to read from the beginning of the file.
open(mycon)
Then when we run readVcf, we will only get the first 1000 SNPs.
first1000 <- readVcf(mycon)
first1000
## class: CollapsedVCF
## dim: 1000 8
## rowRanges(vcf):
## GRanges with 5 metadata columns: paramRangeID, REF, ALT, QUAL, FILTER
## info(vcf):
## DataFrame with 5 columns: AC, AF, DP, AV, NS
## info(header(vcf)):
## Number Type Description
## AC A Integer Allele Count
## AF A Integer Allele Frequency
## DP 1 Integer Total Depth
## AV 1 Integer Average Depth
## NS 1 Integer Number of Samples With Data
## geno(vcf):
## SimpleList of length 2: GT, AD
## geno(header(vcf)):
## Number Type Description
## GT 1 String Genotype
## AD R Integer Allele Depth
If we read again, we would get the next 1000. Here I am assigning it to another object, but depending on what you are doing, this is a great opportunity to use a loop.
next1000 <- readVcf(mycon)
next1000
## class: CollapsedVCF
## dim: 1000 8
## rowRanges(vcf):
## GRanges with 5 metadata columns: paramRangeID, REF, ALT, QUAL, FILTER
## info(vcf):
## DataFrame with 5 columns: AC, AF, DP, AV, NS
## info(header(vcf)):
## Number Type Description
## AC A Integer Allele Count
## AF A Integer Allele Frequency
## DP 1 Integer Total Depth
## AV 1 Integer Average Depth
## NS 1 Integer Number of Samples With Data
## geno(vcf):
## SimpleList of length 2: GT, AD
## geno(header(vcf)):
## Number Type Description
## GT 1 String Genotype
## AD R Integer Allele Depth
Compare to see that they are really different.
rowRanges(first1000)
## GRanges object with 1000 ranges and 5 metadata columns:
## seqnames ranges strand | paramRangeID
## <Rle> <IRanges> <Rle> | <factor>
## MockRefGenome:1731_A/G MockRefGenome 1731 * | <NA>
## MockRefGenome:1851_T/A MockRefGenome 1851 * | <NA>
## MockRefGenome:3311_A/G MockRefGenome 3311 * | <NA>
## MockRefGenome:3833_G/T MockRefGenome 3833 * | <NA>
## MockRefGenome:5102_T/C MockRefGenome 5102 * | <NA>
## ... ... ... ... . ...
## MockRefGenome:189686_C/A MockRefGenome 189686 * | <NA>
## MockRefGenome:190184_C/T MockRefGenome 190184 * | <NA>
## MockRefGenome:190190_T/C MockRefGenome 190190 * | <NA>
## MockRefGenome:190326_G/A MockRefGenome 190326 * | <NA>
## MockRefGenome:190895_C/T MockRefGenome 190895 * | <NA>
## REF ALT QUAL
## <DNAStringSet> <DNAStringSetList> <numeric>
## MockRefGenome:1731_A/G A G <NA>
## MockRefGenome:1851_T/A T A <NA>
## MockRefGenome:3311_A/G A G <NA>
## MockRefGenome:3833_G/T G T <NA>
## MockRefGenome:5102_T/C T C <NA>
## ... ... ... ...
## MockRefGenome:189686_C/A C A <NA>
## MockRefGenome:190184_C/T C T <NA>
## MockRefGenome:190190_T/C T C <NA>
## MockRefGenome:190326_G/A G A <NA>
## MockRefGenome:190895_C/T C T <NA>
## FILTER
## <character>
## MockRefGenome:1731_A/G PASS
## MockRefGenome:1851_T/A PASS
## MockRefGenome:3311_A/G PASS
## MockRefGenome:3833_G/T PASS
## MockRefGenome:5102_T/C PASS
## ... ...
## MockRefGenome:189686_C/A PASS
## MockRefGenome:190184_C/T PASS
## MockRefGenome:190190_T/C PASS
## MockRefGenome:190326_G/A PASS
## MockRefGenome:190895_C/T PASS
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
rowRanges(next1000)
## GRanges object with 1000 ranges and 5 metadata columns:
## seqnames ranges strand | paramRangeID
## <Rle> <IRanges> <Rle> | <factor>
## MockRefGenome:190913_A/G MockRefGenome 190913 * | <NA>
## MockRefGenome:191217_G/A MockRefGenome 191217 * | <NA>
## MockRefGenome:191479_G/A MockRefGenome 191479 * | <NA>
## MockRefGenome:191569_C/T MockRefGenome 191569 * | <NA>
## MockRefGenome:191604_G/T MockRefGenome 191604 * | <NA>
## ... ... ... ... . ...
## MockRefGenome:324994_G/T MockRefGenome 324994 * | <NA>
## MockRefGenome:325077_T/A MockRefGenome 325077 * | <NA>
## MockRefGenome:325099_C/T MockRefGenome 325099 * | <NA>
## MockRefGenome:325368_A/G MockRefGenome 325368 * | <NA>
## MockRefGenome:325552_C/A MockRefGenome 325552 * | <NA>
## REF ALT QUAL
## <DNAStringSet> <DNAStringSetList> <numeric>
## MockRefGenome:190913_A/G A G <NA>
## MockRefGenome:191217_G/A G A <NA>
## MockRefGenome:191479_G/A G A <NA>
## MockRefGenome:191569_C/T C T <NA>
## MockRefGenome:191604_G/T G T <NA>
## ... ... ... ...
## MockRefGenome:324994_G/T G T <NA>
## MockRefGenome:325077_T/A T A <NA>
## MockRefGenome:325099_C/T C T <NA>
## MockRefGenome:325368_A/G A G <NA>
## MockRefGenome:325552_C/A C A <NA>
## FILTER
## <character>
## MockRefGenome:190913_A/G PASS
## MockRefGenome:191217_G/A PASS
## MockRefGenome:191479_G/A PASS
## MockRefGenome:191569_C/T PASS
## MockRefGenome:191604_G/T PASS
## ... ...
## MockRefGenome:324994_G/T PASS
## MockRefGenome:325077_T/A PASS
## MockRefGenome:325099_C/T PASS
## MockRefGenome:325368_A/G PASS
## MockRefGenome:325552_C/A PASS
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
When you are all done reading, close the connection.
close(mycon)
Keeping only specific fields or genomic regions
So far we have read all of the information from the VCF, even though there are some fields that we haven’t looked at at all. The ScanVcfParam function is really useful if we want to save memory by only importing fields that we want.
For fixed fields, let’s ignore QUAL and FILTER since GBS-SNP-CROP didn’t put useful information there. We plan to re-call the genotypes as tetraploid, so we can also drop the diploidized genotypes in the GT field. And in the INFO column, maybe we only care about DP and NS.
myparam <- ScanVcfParam(fixed = "ALT", info = c("DP", "NS"), geno = "AD")
open(mycon)
first1000slim <- readVcf(mycon, param = myparam)
close(mycon)
first1000slim
## class: CollapsedVCF
## dim: 1000 8
## rowRanges(vcf):
## GRanges with 3 metadata columns: paramRangeID, REF, ALT
## info(vcf):
## DataFrame with 2 columns: DP, NS
## info(header(vcf)):
## Number Type Description
## DP 1 Integer Total Depth
## NS 1 Integer Number of Samples With Data
## geno(vcf):
## SimpleList of length 1: AD
## geno(header(vcf)):
## Number Type Description
## AD R Integer Allele Depth
rowRanges(first1000slim)
## GRanges object with 1000 ranges and 3 metadata columns:
## seqnames ranges strand | paramRangeID
## <Rle> <IRanges> <Rle> | <factor>
## MockRefGenome:1731_A/G MockRefGenome 1731 * | <NA>
## MockRefGenome:1851_T/A MockRefGenome 1851 * | <NA>
## MockRefGenome:3311_A/G MockRefGenome 3311 * | <NA>
## MockRefGenome:3833_G/T MockRefGenome 3833 * | <NA>
## MockRefGenome:5102_T/C MockRefGenome 5102 * | <NA>
## ... ... ... ... . ...
## MockRefGenome:189686_C/A MockRefGenome 189686 * | <NA>
## MockRefGenome:190184_C/T MockRefGenome 190184 * | <NA>
## MockRefGenome:190190_T/C MockRefGenome 190190 * | <NA>
## MockRefGenome:190326_G/A MockRefGenome 190326 * | <NA>
## MockRefGenome:190895_C/T MockRefGenome 190895 * | <NA>
## REF ALT
## <DNAStringSet> <DNAStringSetList>
## MockRefGenome:1731_A/G A G
## MockRefGenome:1851_T/A T A
## MockRefGenome:3311_A/G A G
## MockRefGenome:3833_G/T G T
## MockRefGenome:5102_T/C T C
## ... ... ...
## MockRefGenome:189686_C/A C A
## MockRefGenome:190184_C/T C T
## MockRefGenome:190190_T/C T C
## MockRefGenome:190326_G/A G A
## MockRefGenome:190895_C/T C T
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
ScanVcfParam can also be used to subset samples.
myparam2 <- ScanVcfParam(fixed = "ALT", info = c("DP", "NS"), geno = "AD",
samples = c("JM-2014-H-28", "JM-2014-K-8", "JM-2014-M-5", "JM-2014-S-5"))
open(mycon)
first1000slim2 <- readVcf(mycon, param = myparam2)
close(mycon)
first1000slim2
## class: CollapsedVCF
## dim: 1000 4
## rowRanges(vcf):
## GRanges with 3 metadata columns: paramRangeID, REF, ALT
## info(vcf):
## DataFrame with 2 columns: DP, NS
## info(header(vcf)):
## Number Type Description
## DP 1 Integer Total Depth
## NS 1 Integer Number of Samples With Data
## geno(vcf):
## SimpleList of length 1: AD
## geno(header(vcf)):
## Number Type Description
## AD R Integer Allele Depth
geno(first1000slim2)$AD[1:10,]
## JM-2014-H-28 JM-2014-K-8 JM-2014-M-5 JM-2014-S-5
## MockRefGenome:1731_A/G Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:1851_T/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:3311_A/G Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:3833_G/T Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:5102_T/C Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:5104_C/G Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:5127_G/A Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:5327_A/G Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:5498_A/C Integer,2 Integer,2 Integer,2 Integer,2
## MockRefGenome:6284_G/A Integer,2 Integer,2 Integer,2 Integer,2
A third handy use for ScanVcfParam is specifying a genomic region with the which parameter. The one catch is that you can’t use it with TabixFiles that have a yieldSize other than NA. Here is how I would read from 10000 to 11000 bp on MockRefGenome, assuming it were a real chromosome.
myparam3 <- ScanVcfParam(which = GRanges("MockRefGenome", IRanges(10000, 11000)))
specific_range <- readVcf(mybg, genome = "MockRefGenome", param = myparam3)
rowRanges(specific_range)
## GRanges object with 2 ranges and 5 metadata columns:
## seqnames ranges strand | paramRangeID
## <Rle> <IRanges> <Rle> | <factor>
## MockRefGenome:10163_C/G MockRefGenome 10163 * | <NA>
## MockRefGenome:10168_C/A MockRefGenome 10168 * | <NA>
## REF ALT QUAL
## <DNAStringSet> <DNAStringSetList> <numeric>
## MockRefGenome:10163_C/G C G <NA>
## MockRefGenome:10168_C/A C A <NA>
## FILTER
## <character>
## MockRefGenome:10163_C/G PASS
## MockRefGenome:10168_C/A PASS
## -------
## seqinfo: 1 sequence from MockRefGenome genome; no seqlengths
ScanVcfParam is not only useful for importing data, but also filtering.
filter3_file <- "Msa_GSCdemo_filter3.vcf"
filterVcf(mybg, genome = "MsaMockReference",
destination = filter3_file,
prefilters = FilterRules(list(NotAllHet)),
filters = FilterRules(list(DPbelow1000, NoMissing)),
param = myparam2)
cat(readLines(filter3_file, 20), sep = "\n")
## ##fileformat=VCFv4.2
## ##fileDate=20190425
## ##phasing=partial
## ##source=GBS-SNP-CROP
## ##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele Count">
## ##INFO=<ID=AF,Number=A,Type=Integer,Description="Allele Frequency">
## ##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
## ##INFO=<ID=AV,Number=1,Type=Integer,Description="Average Depth">
## ##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data">
## ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
## ##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allele Depth">
## ##contig=<ID=MockRefGenome,assembly="MsaMockReference">
## #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT JM-2014-H-28 JM-2014-K-8 JM-2014-M-5 JM-2014-S-5
## MockRefGenome 92975 MockRefGenome:92975_C/A C A . . DP=982;NS=8 AD 92,0 143,0 40,19 134,33
## MockRefGenome 93207 MockRefGenome:93207_C/G C G . . DP=965;NS=8 AD 114,0 144,40 60,0 123,42
## MockRefGenome 95964 MockRefGenome:95964_C/T C T . . DP=955;NS=8 AD 99,0 115,17 92,0 154,0
## MockRefGenome 98167 MockRefGenome:98167_C/A C A . . DP=889;NS=8 AD 0,32 69,31 52,0 42,0
## MockRefGenome 101036 MockRefGenome:101036_G/T G T . . DP=966;NS=8 AD 137,0 140,0 71,0 121,44
## MockRefGenome 101076 MockRefGenome:101076_G/C G C . . DP=967;NS=8 AD 137,0 140,0 72,0 121,44
## MockRefGenome 103076 MockRefGenome:103076_C/A C A . . DP=961;NS=8 AD 83,0 217,0 76,0 146,0